定量核磁分析看这一篇就足够了!
各种不同的分析、分离纯化和结构鉴定技术应用于药物研发的整体过程,其中对化合物的纯度确定是分析工作的重要内容之一。在新药研发过程中,许多待标定的化合物是新化合物,没有已知纯度的自身标准品。这时,需要经验丰富的分析人员对化合物进行各种不同的测试,主要包括HPLC纯度、有关物质、杂质、水分、残留溶剂、无机盐等等,然后计算待测化合物的纯度。这个传统的物料平衡法需要的样品量较大,并且非常耗时。
近些年来,利用定量核磁技术对化合物的纯度进行标定得到了越来越广泛的应用。定量核磁的原理是什么?具体的操作流程如何?在药物研发中具体有哪些实际应用?下面就这些问题做了详细的讲解。
定量核磁(QNMR)的原理
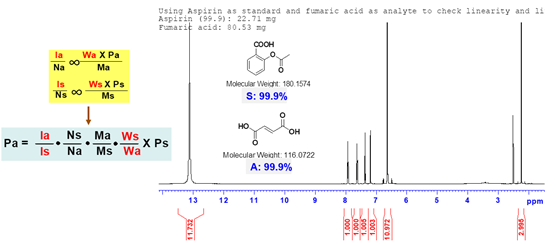
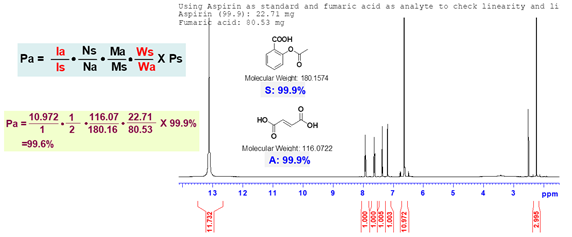
定量核磁灵敏度高,易操作
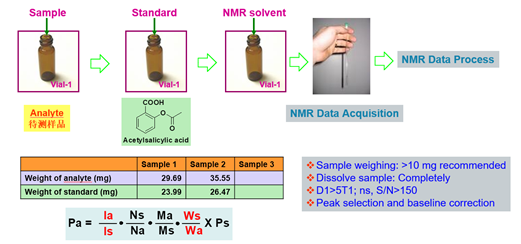
定量核磁在药物研发中的应用案例
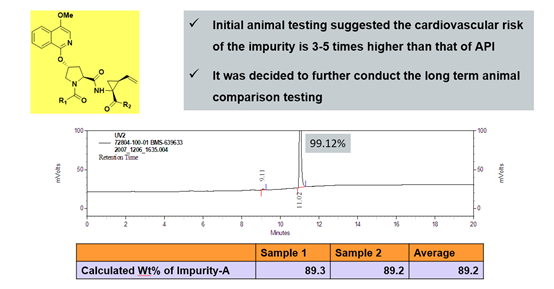
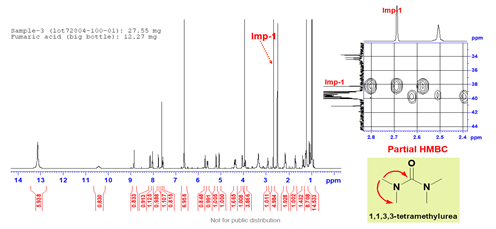
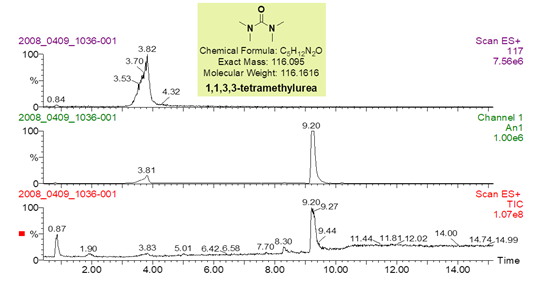
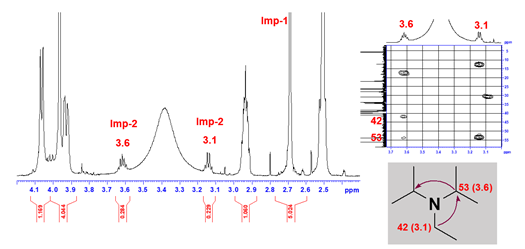
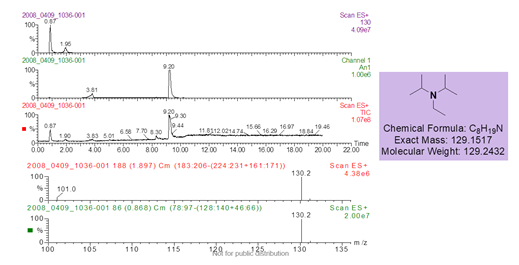
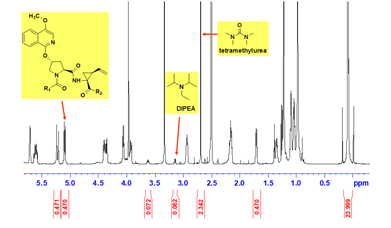
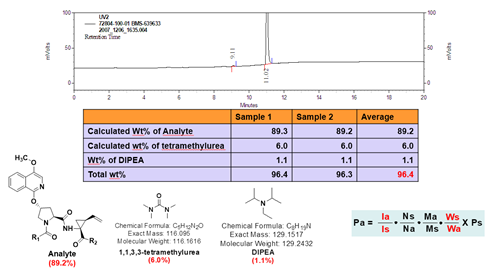
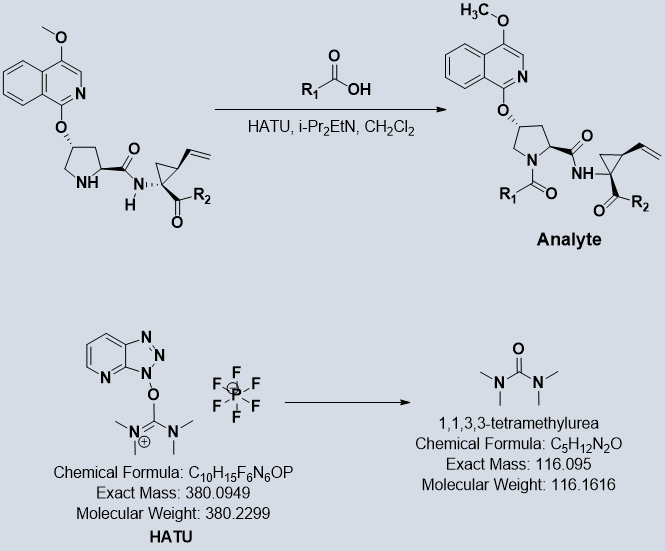
定量核磁的优点

展源
何发
相关文章
-

QC, IQC, IPQC, QA,到底是什么鬼?
2020-05-27
-

AAS法分析茶叶中的铅,镉,砷
2020-05-27
-

称量时增量法与减量法辨析
2022-07-20
-
红外光谱分析,你了解多少?
2021-01-11
-

三聚氰胺,你还要害多少人
2020-05-27
-

HPLC检测器,你了解吗?
2024-03-06
-

超净工作台原理,使用与维护
2020-05-27
-

核磁知识清单!
2020-12-18
-

选对色谱柱,快速开发方法
2020-05-27

加载更多