气相色谱定量方法
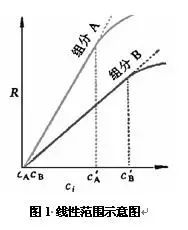
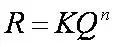
gi=ms/Ai
式中ms是标准样品中组分i的含量,Ai是标准样品谱图中组分i的峰面积。
2.外标法的计算公式
mi=Ai * gi
这里mi是未知样品中组分i的含量。
Gi=gi/gs
式中gi是组分i的绝对校正因子,gs是标准物质的绝对校正因子。
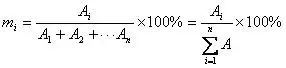
当各个组分的绝对校正因子不同时,可以采用带校正因子的面积归一化法来计算。事实上,很多时候样品中各组分的绝对校正因子并不相同。为了消除检测器对不同组分响应程度的差异,通过用校正因子对不同组分峰面积进行修正后,再进行归一化计算。其计算公式如下:
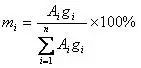
与面积归一化法的区别在于用绝对校正因子修正了每一个组分的面积,然后再进行归一化。注意,由于分子分母同时都有校正因子,因此这里也可以使用统一标准下的相对校正因子,这些数据很容易从文献得到。
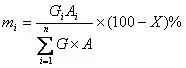
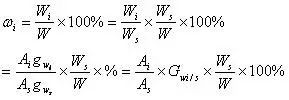
式中, Ai,As分别为待测组分和内标物的峰面积;Ws,W分别为内标物和样品的质量;Gwi/s是待测组分对于内标物的相对质量校正因子(此值可自行测定,测定要求不高时也可以由文献中待测组分和内标物组分对苯的相对质量校正因子换算求出)。
内加法需要除了和内标法一样进行一份添加样品的处理和分析外,还需要对原始样品进行分析,并根据两次分析结果计算得到待测组分含量。和内标法一样,内加法对进样量并不敏感,不同之处在于至少需要两次分析。下面我们用一个实际应用的例子来说明内加法是如何工作的:
题:在分析某混合芳烃样品时,测得样品中苯的面积为1100,甲苯的面积为2000,(其它组分面积略)。精确称取40.00g该样品,加入0.40g甲苯后混合均匀,在同一色谱仪上进混合后样品测到苯的面积为1200,甲苯的面积为2400,试计算甲苯的含量。
分析:本题的分析过程是一个典型的内加法操作,其中内加物为甲苯,待测组分为甲苯和苯。
解:1. 由于进样量并不准确,因此两次分析的谱图很难直接进行对比。为了取得可以对比的一致性,我们通过数字计算调整两次分析苯的峰面积相等。此时由于两次分析苯峰面积相等,因此可以断定两次分析待测样品的进样量是相等的。需要注意的是:此时两次分析的总的进样量并不相等,添加后样品比原始样品调整后的进样量中,多了添加的内标物的量。
调整可以用原始样品谱图为依据,也可以用添加后样品谱图为依据。但是通常采用原始样品作为依据以便计算最终结果时比较简单。注意:选用的依据不同,中间计算结果会产生差异,但不会影响最终结果。依据的谱图一旦选定,计算就应该围绕此依据进行。
在以原始样品谱图为依据的情况下,调整添加后样品谱图中甲苯的峰面积如下:
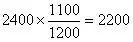
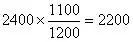
据此可以计算得到在以原始样品谱图为依据的条件下甲苯的绝对校正因子g:
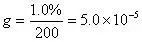
此时,可以根据外标法,以原始样品谱图为依据,计算得到甲苯的含量为
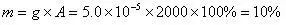
答:此样品中甲苯的含量为10%。
另:通过相对校正因子,容易得到苯的校正因子并计算得到苯的含量。
2、归一化法不要求精确进样量,但要求所有组分都必须出峰,或者所有出峰组分的总含量已知。有些时候虽然能够精确进样量,但所有组分都出峰的情况下,也使用归一化法。因为此时归一化法相当于外标法定量后,对总量进行归一化误差修正。
3、内标法是无法精确进样量、不是所有组分都出峰的情况下,解决定量的办法。相对而言,操作和计算都很复杂。内标法的关键是要能够找到合适的内标物。内标法的称量误差,应小于色谱正常定量分析误差。
4、再无法找到合适内标物的无奈情况下,可以使用内加法。内加法操作复杂,计算繁琐,不是一种常用的定量方法。
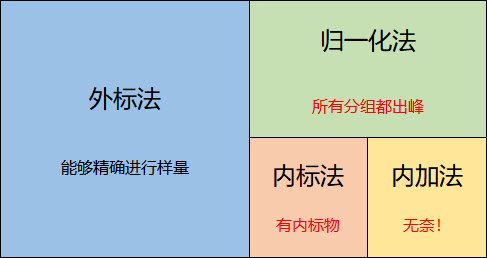
图2 各种定量方法的关系
展源
何发
相关文章
-

检测有机氯类农药,气相色谱法检测法
2021-01-12
-

选对色谱柱,快速开发方法
2020-05-27
-

气相色谱法测定苍术中茅术醇含量
2020-05-27
-

气相色谱法测定心力丸中麝香酮的含量
2020-10-09
-

AAS法分析茶叶中的铅,镉,砷
2020-05-27
-

气相色谱法之保留时间
2021-09-21
-

毛细管气相色谱法测定止咳枇杷颗粒中薄荷脑的含量
2020-10-09
-

气相色谱分析方法的建立步骤
2023-11-23
-

气相色谱法检测温室气体
2020-05-27

加载更多