农药残留的多元分析法
农药残留的多元分析法

农药分析在植物保护方面起着很重要的作用。如今,已知有超过1000种物质需要被检测。但是,这些农药的物理、化学性质的不同,使得通用检测方法的开发遇到了一些困难。而快速、动态的仪器使得采用快速、简便的样品制备及检测的多元方法成为可能。本文介绍了已在Eurofins 苏州实验室应用的最新分析方法,在一个分析过程中可以检测上百个化合物。另外,现代多元分析方法的局限性也表明,由于化合物的化学、物理性质差异以及仪器设备的局限,还无法实现对所有可能的分析物进行检测。
自公元前2500年,人类开始使用农药来保护作物。最早的农药是4500年前在Sumeria使用的硫。15世纪,有毒化学品如砷、汞和铅被用来除掉作物中的害虫。17世纪,从烟叶中提取出来的硫酸烟精被用作杀虫剂。19世纪,出现了两个天然杀虫剂,一个是由菊花中提取出的除虫菊,另一个是从热带蔬菜根中提取出的鱼藤酮。
1939年,德国化学家Paul Müller发现了DDT是非常有效的杀虫剂,标志着现代杀虫剂的开始使用。很快,这种杀虫剂便风靡全球。
但人们并不愿意看到农药在食品中的残留,对于其毒性的关注,有关农药残留的分析很早就开始了。最早的官方农药检测实验室于21世纪50年代在英国建立,在随后的几十年中,欧洲其它国家也相继建立了农药检测实验室。最初是采用生物学检测,用果蝇与杀虫剂相接触。当果蝇暴露在杀虫剂环境中,提高杀虫剂剂量,就可以观察到果蝇中毒和死亡的现象。
起初研发出的是单残留的测定方法,即一个方法只能一次分析一种农药。而太多活性物质的存在,激发了人们去开发一个适合的多元方法,同时检测大量被筛选的化合物。值得一提的第一个方法是由化学家P.A. Mill于1960年代开发的。首个德国的官方方法颁布于1968年,即采用薄层色谱法半定量地同时测定6个有机氯农药。通过在紫外光下观察硅胶板上的斑点大小及强弱来判断化合物浓度的大小。
后来,随着配有电子捕获(ECD)检测器和氢火焰(FID)检测器的气相色谱仪的出现,在上世纪60年代后期和70年代初期,很多测定多种农药残留的方法被开发出来。但由于这些检测器的选择性不是足够好,因此需要花费大量的精力和时间进行样品的净化与分离,还需要采用大量的柱色谱技术来处理样品,以使样品可以适合最终的仪器检测。此外,这些方法还存在一个问题即不能检测大多数的极性物质。因此,新方法开发的关键就在于对更大极性的物质如有机磷和有机氮化合物的涵盖。其中一个方法是采用丙酮提取,然后用液-液萃取技术将水除掉,通过在水层中加入氯化钠,显著提高在萃取过程中极性化合物的收率。
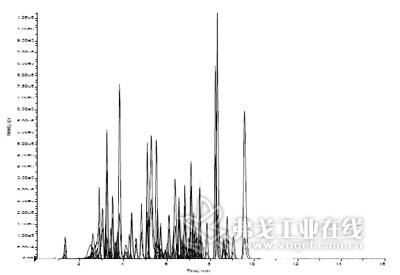
图1. 80个化合物对照片在LC-MS/MS中同时被检测,浓度为10 ppb。
这一方法成为当时的官方方法,在美国颁布于AOAC 985.22,德国为DFG S-19,并在全世界范围内广泛应用。对于提取得到的物质,采用凝胶渗透色谱和柱色谱可以进行进一步的分离纯化,得到干净的待测样品。最早采用配有ECD 和FPD检测器的气相色谱仪对很多有机氯和有机磷农药进行检测,随着质谱方法的使用,几乎所有含氮的有机农药都可以被检测。尽管这些方法已经很陈旧了,但仍然在世界范围内广泛地被使用。
在21世纪80年代,人们投入了大量的精力来进一步开发上述的官方方法,但是,其中一些方法主要的劣势仍无法被克服。主要包括以下几方面:
溶剂消耗量大;
样品净化过程过于复杂、耗时;
水溶性物质常常在样品净化过程中丢失;
新型的农药不适合用气相色谱(GC)分析-或GC检测无法满足要求。
而且,食物常常容易变质,在检测报告出来很久之前食品常常已经被消耗掉了,故耗时长的检测过程不太会被采用。
此时,一个巨大的飞跃是成功地将液相色谱和质谱技术联用。自从21世纪90年代开始,这种方便的实验技术便被应用到日常分析中;使用选择性和灵敏度高的质谱(MS)检测农药并不只局限在气相色谱方法中。此外,串联的质谱(MS/MS)的应用开启了检测器灵敏度的新纪元。使用3个质量连续的质谱仪,用于定性和定量的特定离子(碎片离子)即可被测定。高扫描速率使得一次运行可进行多种化合物的测定成为可能。就在最近,这种方法被认为是欧洲农药残留测定的标准方法。
最新的方法不仅把LC和MS/MS连用起来;串联质谱还与GC相结合,使用这种复杂的色谱技术来进行日常的农药分析。
农药分析的需求和设想
对于实验室来说,农药分析所面临的挑战是非常大的。以下列出了几点主要要求:
可检测的物质范围宽(越多越好);
最低定量限(LOQ)要低,需符合官方所规定的最大残留要求;
检测周期短及快速的服务;
消费者可以负担的检测费用;
环境方面:溶剂及化学品消耗少,少用或避免使用有毒化学物质;
高质量分析(准确度及精密度),没有用户愿意接受错误的结果;
可适用于不同类型的样品。
在过去的几十年中,无数的方法被开发出来却又消失了,其原因就是忽视了以上所述的要求。接下来,我会阐明在实验室中如何实现上述要求。

图2. 新型的LC-MS/MS仪器。
简便的样品制备
由于食物基质非常复杂,无法用一种通用的方法来净化。而干扰检测的一些成分又必须要在净化过程中去掉,如果仪器足够好的话,可以将样品进行稀释,使得基质效应被抑制或者不影响检测。对于基质效应不强的待测物,现代的多元方法可采用简单、快速的净化步骤,但在基质效应很强的情况下,样品纯化过程就不得不延长。
如今,实验室已不再使用大型的柱色谱纯化手段。在一个快速有效的提取步骤之后,净化过程被缩减到最小,有时完全被省略或者使用小的更易于操作的固相萃取(SPE)柱来完成。
最小化
传统方法的样品使用量约100g,为了提取完全,需消耗大量的溶剂。之后,提取液需要被浓缩,再进行下一步样品的净化。而现代方法大大减少了样品的用量,并保持很小的溶剂/样品量比值,溶剂的使用量和样品提取液浓缩的步骤也大大减少。实验人员只需要处理小量的样品和溶剂,速度更快,更简便且更安全,并达到保护环境的目的。
自动化
如今日常的实验室作业就像工厂一样,大量样品被收集起来然后同时平行地被处理,实验人员可以专注于他正在处理的一批样品来维持一个高质量的操作。另外,数据分析是整个过程中的关键点。使用多元分析方法后,许多化合物包括样品的活性成分在不同的浓度水平被检出,许多色谱图要被分析处理。为保持对大量信息的汇总,迫切需要一个正确的定性和定量方法。
自动化的例子:
使用样品分离设备会有利于盐类混合物样品的制备;
样品浓缩过程可以通过不同体积的平行蒸发系统来加速;
分析模块软件可以把所有样品/化合物的原始数据放入一个文件内。所有有用的数据信息都可以一次被分析。实验人员可以从定性鉴定、线性、回收率及定量这些数据来立即判断是否符合标准要求。
质量保证
一个广泛被认可的质量标准指导性文件DG-SANCO(健康和消费者总署)描述了方法验证和分析质量控制的要求,以保证数据的准确性。主要的目标是保证分析结果的质量和可比性,确保结果的准确性,避免假阳性及假阴性。好的实验室大都采用这一方法,一方面可验证其农药的分析方法,另一方面可进行日常的样品定性和定量检测。
化合物的图谱
农药分析实验室在提供源源不断的化合物目录方面互相竞争,超过500种的目录也并不少见。能够搜集这么多化合物目录的主要原因是得益于强大的质谱分析,这种检测器不仅提供了选择性也提供了高灵敏度。传统的检测器在某一保留时间处只产生一个信号,但对于定性的鉴别还需要进行另外的分析过程。此外,对于正确的定量分析而言,首先,分析方法必须保证所有化合物均被有效分离。凭借质谱分析,保留时间和被测量的质量数(或者碎片离子)可以实现被分析物的定性。另外,化合物的完全分离并不是定量的先决条件,一个无干扰的质量数足够用于定量。快速扫描的质谱仪器能够在一次运行中同时检测几百个不同的化合物,几毫秒的保留时间也正适合于在这样高灵敏度的前提下将所测定的分析物记录下来。
农药分析的局限性
但现在还不可能建立一个方法能一次分析上千个化合物,在分析方法上仍存在一些无法逾越的障碍。
化学和物理特性
不同种农药化合物有着非常不一样的化学结构。现在还不能找到一个合适的提取和净化方法既能够得到所有的活性化合物,又有满意的回收率。多元方法经常是提取效率和净化效果的折中,并使得所有待检测的化合物得到令人满意的准确度、精密度。
基质效应
在样品提取时,随被分析物一起被提取出来的共提取物常常会干扰我们的分析。通常大部分的共提取物会在样品制备过程中被除掉或者被稀释,但是剩余的部分仍会对被分析物的信号形成干扰(抑制/增强),并造成错误的结果。通过加入内标物(同位素标记物质)可以校正共提取物的影响,但是这种方法在多元方法中的使用很有限。
仪器设备的局限
一方面有色谱部分的局限,另一方面测量时间也是一个限制因素。所有的化合物都需要在一个合理的时间内以合理的色谱峰形顺序被洗脱出来,由于每一个色谱峰的色谱行为均不同,因此,这对农药分析来说并不容易。另外,许多离子和碎片离子需要在同一时间被一一测量。通过减少对被测量离子(碎片离子)的扫描时间,被记录下来的化合物峰的数量也会增加。然而,如果扫描时间太短了,灵敏度就会大大降低。
总结和预测
农药分析在近50年里经历了飞速发展。现代样品制备技术和精密仪器的使用使得一次运行分析几百种活性物质成为可能。由于最新仪器所具有的高灵敏度,大部分农药的最低定量限(LOQ)可以达到10 ppb。对于快速分析方法,样品的净化更易于掌握,对于基质效应比较强的样品,则需要另外的纯化处理。如今所采用的方法检测周期都较短,所以24h快速的服务也变得可能。
在仪器开发方面,灵敏度的提高更加重要,尤其是当更多GC-MS/MS仪器投入使用时。由于检测灵敏度的提高,样品液会被进一步稀释,这样基质效应的影响则会被减少。扫描速度更快的质谱仪可以对更多的化合物进行定性鉴定,农药分析的化合物目录也因此会变得更长。
欧陆分析技术服务(苏州)有限公司
展源
何发
热点文章
-

硫酸稀释是酸入水还是水入酸?
2026-04-23
-

【收藏】标准溶液的配制和计算
2026-04-07
-

【必看】红外光谱仪结构图与原理
2026-04-28
-

【收藏】常用溶液配制速查
2026-04-10
-

53种常用指示剂配制方法
2026-04-14
-

珀金埃尔默半导体行业检测解决方案Brochure
2026-05-13
-

滴定管到底该怎么润洗?
2026-04-14
-

独立百天,新生启航:Solstice Advanced Materials中国首秀,锚定先进计算、可持续发展、生命科学新航道
2026年1月末,距离从霍尼韦尔正式分拆独立,并在纳斯达克挂牌上市将满百日时,全新的特种材料公司 Solstice Advanced Materials在上海举办了其独立运营后的首次中国媒体见面会。
作者:
-
安捷伦在中国:加速本土创新与绿色发展的双重推进
-
洞察未来,共谋发展 “ 数·智·未来 ” 安捷伦未来实验室媒体圆桌会成功举办
-
食品检验理化常用国家标准与要点
-
水分测定方法开发研究&检测相关问题故障分析解决解读
-
药物常用的晶型表征方法

评论
加载更多