仿制药中有关物质研究(连载二)
质量标准的制订
杂质和辅料的定位
对于已知降解 杂质A和B,通常采用对照品法或相对保留时间法,前者需制备对照品并供日常检测用,后者简便易行,故推荐后者。检测制剂时有时会出现辅料峰,保留时间通常 较短,可采用“扣除主成分峰相对保留时间多少倍前的辅料峰”的办法;如辅料峰位于中间位置,则只能在质量标准中规定:取某辅料、配制成某浓度进样测定,该 辅料峰不计入的办法。
杂质定量法
由于杂质与主成分结构式不同、紫外吸收不同,故在同一波 长下采用峰面积代表含量时需引入校正因子。针对杂质A和B的校正因子,如文献报道值已翔实确凿,则可直接采用;如怀疑或不存在报道值,则采用液质联用仪或 其他检测手段,甄别出该两杂质结构式,随后从国外药典会或某实验室购买来测得该杂质校正因子;或通过自我合成制备 → 结构式确认→ 获得纯度值 → 测得校正因子。这里需提醒的是:无论何种途径获得的杂质对照品,纯度值无需很高,只要该值准确、误差范围以内即可。针对特有杂质E和F,也是遵循以上方法 获得校正因子后计算含量。通常,校正因子在0.9~1.1时可忽略,超出0.5~5.0时应考虑改变检测波长,如无法调节,应考虑采用杂质对照品法。
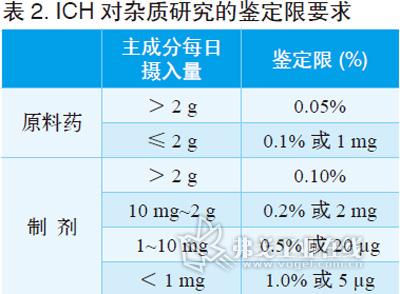
杂质限度的规定
由于杂质A和B是主成分降解杂质,故其限度值在既有标准应有规定,参照即可其他杂质无需再参照既有质量标准,可自行拟定为:其他最大单个未知杂质不得过 0.5%,其次单个未知杂质不得过0.2%,所有杂质不得过1.5%。针对五类改剂型的研发,当用药途径一致时,仍可参照该限度制订;若用药途径发生改 变,则应具体分析。
系统适用性试验
系统适用性试验除在方法开发时用到,在日常检测中也应被使用,从而保证即时的色谱条件可满足测定需要。英国药典在所有液相法检测有关物质时均有此项规定。通常可采用如下方法。
使用与主成分最难分离的杂质;
如知晓原料药合成中间体G是最紧邻主成分峰前的杂质,可将其拟定入系统适用性试验中,只要保证该杂质与主成分峰的分离,便可确保其他杂质的分离;
使用已知杂质对照品;
如采用对照品法测定杂质A,则可规定该杂质峰与主成分峰具有适当的分离度,以保证位于中间的杂质B与G能与主成分峰分开,而不能仅规定1.5的分离度。
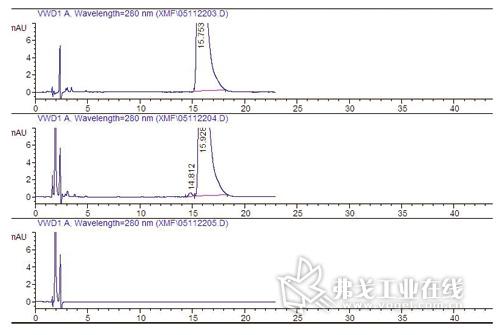
图3. 系统适用性强破坏试验图谱(下/空白溶剂;上/未破坏的供试品溶液;中/氧化破坏试验:规定产生一主成分峰前的杂质峰,且分离度应符合规定)。
采用破坏试验
如主成分在某强破坏试验下较易产生杂质,且该杂质又在质量标准中有所规定限制,则可采用,如图3所示。不建议仅规定主成分峰柱效、拖尾因子等作法,这些参数无法直接表达出主成分与杂质的分离效果。
复方制剂的研究
研究思路同前,仍是主要关注降解新增杂质,来源可能为:各原料间相互作用、原料与辅料间相互作用、原辅料与包材间相互作用等。不同之处如下。
色谱系统
研究阶段,通过确定色谱柱型号加以区域性分离、明确各杂质归属后进行检测的方法最为常用,且当一套色谱系统难以将各主成分与杂质均分离时,可建立两套/多套 色谱系统进行测定。如色谱系统一测定主成分A及其杂质,色谱系统二测定主成分B与其杂质,并验证在该各自系统条件下彼此互不干扰。
根据研究结果制订质量标准
如经以上研究,所有杂质均未增加/变化,则可在质量标准中不予制订有关物质检查项,该思路对于氨基酸等大复方制剂更为适用。如仅主成分A的一个已知杂质不断增加,则质量标准中仅制订该杂质测定法与限度。
很多研究者由于未充分理解制剂质量标准仅关注降解杂质的理念,将所有杂质均订入了质量标准。不变化的杂质由于已在原料药/辅料中加以了制御,故制剂质量标准中便无需再控制。
实验与分析
展源
何发
热点文章
-

硫酸稀释是酸入水还是水入酸?
2026-04-23
-

【收藏】标准溶液的配制和计算
2026-04-07
-

【必看】红外光谱仪结构图与原理
2026-04-28
-

【收藏】常用溶液配制速查
2026-04-10
-

53种常用指示剂配制方法
2026-04-14
-

珀金埃尔默半导体行业检测解决方案Brochure
2026-05-13
-

滴定管到底该怎么润洗?
2026-04-14
-

独立百天,新生启航:Solstice Advanced Materials中国首秀,锚定先进计算、可持续发展、生命科学新航道
2026年1月末,距离从霍尼韦尔正式分拆独立,并在纳斯达克挂牌上市将满百日时,全新的特种材料公司 Solstice Advanced Materials在上海举办了其独立运营后的首次中国媒体见面会。
作者:
-
安捷伦在中国:加速本土创新与绿色发展的双重推进
-
洞察未来,共谋发展 “ 数·智·未来 ” 安捷伦未来实验室媒体圆桌会成功举办
-
食品检验理化常用国家标准与要点
-
水分测定方法开发研究&检测相关问题故障分析解决解读
-
药物常用的晶型表征方法

评论
加载更多