液质联用法测定奶粉中三聚氰胺总结
液质联用法测定奶粉中三聚氰胺总结
三鹿奶粉事件过去,现在差不多已经冷下来了,我把这几个月做三聚氰胺的体会总结了下,为以后的应急工作提供点参考吧。
07年发生的宠物饲料三聚氰胺事件时,我也注意到了,但因为想到饲料跟我们系统不可能有关系,也就没去管它。奶粉事件严格来说,是质检部门的事,与我们系统并没有关系,但我们这个系统和国内大部分部门没什么差别—好表现,有露面的机会领导是不会错过的,因此在奶粉事件经媒体曝光第二天,领导的命令就来了,并顺便送来了5罐不同批号的三鹿奶粉。而这个任务也就毫无悬念地落到了作为负责液质联用仪和新项目开发的我的头上。接到命令的时候我还在外面开会,而且对三聚氰胺的性质检测方法一无所知。但命令来了,就得立马往回赶,同时马上电话联系借标准(买是来不及了)多亏了07年的饲料事件,很顺利地从本地出入境部门借到标准,马上派学生去拿。我回到单位时,标准已经拿回来,马上上网查资料、配标准、摸质谱条件、样品处理。在中秋假的前一天,从下午4点开始到晚上10点测完样品,总算没牺牲我的中秋假期。中秋假后再回来将方法优化好,当然检测的样品粗算一下也超过了700份,幸亏当时我手下带了两个学生,样品前处理就交给了他们,真是辛苦他们了!
下面主要说说我用液质联用法测定的体会,HPLC法和GC-MS法没试过,不敢在此胡说。个人体会,有不对的地方,欢迎大家指正。
从开始参考的是国内某期刊测饲料的方法到最后用FDA方法(08年10月8日颁布),中间试过很多样品处理方法,具体如下:
1、用1%三氯乙酸或乙腈水(50+50)提取,MCX小柱净化,行不通,部分样品蛋白质沉淀不下来,甚至同一样品,也有的能产生沉淀,有的沉不了。部分样品浑浊,固相萃取根本没法进行,液体在小柱上滤不动。
2、用0.1%三氯乙酸提取后直接进样,因为样品脏,加大稀释倍数,峰形不太好,但可以定量。同时发现随着三氯乙酸浓度增大,峰形变坏越严重。而且因样品未经任何净化手段,离子源用不了多久就很脏了。
3、用0.1%三氯乙酸提取,乙酸锌沉淀蛋白后直接进样,峰形不错,但回收率不稳定。
3、用5%三氯乙酸提取,MCX小柱净化,样品干净,峰形好,但所有样品包括空白奶粉样品也有三聚氰胺峰的出现,换了几个品牌的小柱都一样,计算下来,有零点几ppm,虽然结果不高,但感觉肯定不好。曾与某品牌耗材的技术工程师讨论过这个问题,他解释是自然界三聚氰胺污染普遍,加上液质检测灵敏度高,造成的这种情况。但事实证明这只是他的臆测,我没过柱的样品空白为什么没有峰呢?
4、FDA方法:0.3%甲酸提取后,取100ml加900ml乙腈,混合后出现蛋白质的沉淀,离心过滤后测定,结果好,峰形好,结果稳定。有人以为样品没经固相萃取净化肯定会很脏,但实际应用时没有出现这种情况,做N多份样品后,离子源还很干净。这个是我个人觉得最满意的方法,检验检疫科学院组织的三聚氰胺考核用的就是这个方法,后面的鸡蛋三聚氰胺事件也是用的这个方法。采用工作曲线,根本不用考虑回收率的问题,这个方法最大的好处是可以同时测定三聚氰酸和氰尿酰胺。
总结:刚开始接触时,以为三聚氰胺是个很简单的项目,质谱条件很容易做,单一项目也不担心液相色谱分离的问题,虽然在C18柱上保留差,但改用HILIC柱就一点问题也没有。一做才发现并非如此,三聚氰胺是我到目前为止所遇到项目中基质效应最严重的项目,标准曲线必须用空白样品加标的工作曲线,常用的固相萃取净化方法会带来干扰,三聚氰胺在日常生活接触不少,我们用的很多塑料制品就是三聚氰胺塑料,这些都可能会带来污染;看不少文献提到检测结果有假阳性、假阴性的问题,本人在摸索方法时也有遇见,不过改用FDA方法后,尚未发现出现这种情况。
下面附上本人实际检测的MRM质谱图。
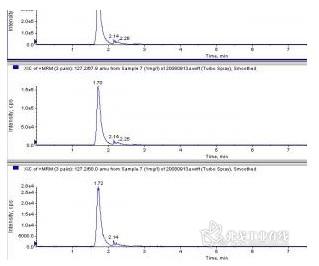
0.1%三氯乙酸提取后直接进样,waters Atlantis C18柱,主峰后面有个小峰,但不影响测定。
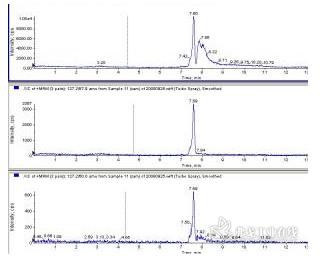
1%三氯乙酸提取后直接进样,HILIC柱,峰前延,定量离子峰后面有个强度不小的干扰峰。
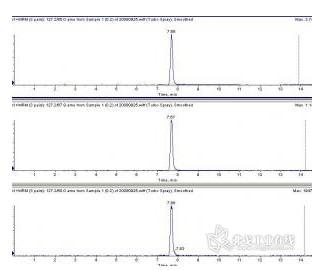
0.1%三氯乙酸提取后,乙酸锌沉淀蛋白后进样,HILIC柱,峰形不错。
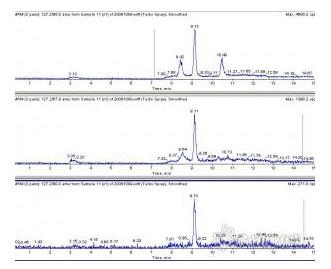
用5%三氯乙酸提取,MCX小柱净化,空白奶粉样,出现了干扰峰。
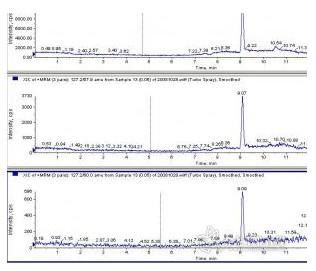
FDA方法
实验与分析
展源
何发
热点文章
-

硫酸稀释是酸入水还是水入酸?
2026-04-23
-

【必看】红外光谱仪结构图与原理
2026-04-28
-

赛默飞ASMS 2026调研有礼
2026-06-15
-

珀金埃尔默半导体行业检测解决方案Brochure
2026-05-13
-

常规玻璃器皿标准洗涤流程
2026-05-27
-

半导体化学机械抛光(CMP)技术
2026-05-12
-

【避坑】内切酶实验四个常见问题大盘点!
2026-05-08
-

独立百天,新生启航:Solstice Advanced Materials中国首秀,锚定先进计算、可持续发展、生命科学新航道
2026年1月末,距离从霍尼韦尔正式分拆独立,并在纳斯达克挂牌上市将满百日时,全新的特种材料公司 Solstice Advanced Materials在上海举办了其独立运营后的首次中国媒体见面会。
作者:
-
安捷伦在中国:加速本土创新与绿色发展的双重推进
-
洞察未来,共谋发展 “ 数·智·未来 ” 安捷伦未来实验室媒体圆桌会成功举办
-
食品检验理化常用国家标准与要点
-
水分测定方法开发研究&检测相关问题故障分析解决解读
-
药物常用的晶型表征方法

评论
加载更多